Running evSeq
Post Installation
Running example data
To confirm that evSeq has been installed and works on your machine, we have provided test datasets that work with the example refseqs. You have three options for running this:
- via Jupyter Notebook
A demo notebook can be found in examples/8-full_demo.ipynb which effectively runs evSeq via the command line. This is also rendered as documentation here. The end of the notebook uses the outputs of this run and compares them to the expected output given the provided example data.
- via Command Line
General info on running evSeq from the command line can be found below.
First, you will need to activate your conda environment (if using it) with
conda activate evSeq
To run the example files, first navigate to the examples folder of the evSeq repository:
cd path/to/evSeq/examples
From here, run evSeq as follows:
evSeq refseqs/DefaultRefSeq.csv ../data/multisite_runs
This should start the run and create an evSeqOutput folder in the current working directory (evSeq/examples) with a timestamped results folder. Once the run has finished, confirm it has done so without errors (they will be sent to the standard output and the log file) and compare the results to the expected results in evSeq/data/multisite_runs/evSeqOutput/expected. You can find example code for comparing these in the demo documentation in the above section, using the function compare_to_expected from evSeq.util.
- via GUI
General info on running evSeq from the GUI can be found below.
First, double click the evSeq shortcut on the Desktop (if you have not moved it to another location). This will open the GUI; it may take a minute, especially the first time opening it.
In the GUI, click on the refseq selector and navigate to the examples folder (path/to/evSeq/examples) and select DefaultRefSeq.csv.
For folder, navigate to the data folder (path/to/evSeq/data) and select multisite_runs.
Then click Start.
This should start the run and create a new timestamped results folder in the evSeqOutput directory already present in multisite_runs folder (the same location as the folder argument). Once the run has finished, confirm it has done so without errors (they will be sent to the GUI console and the log file) and compare the results to the expected results in evSeq/data/multisite_runs/evSeqOutput/expected. You can find code for comparing these in the demo documentation in the above section, or do so visually by comparing the Platemaps.html files.
Troubleshooting
For common problems encountered when using evSeq, please reference Troubleshooting.
Known Limitations
evSeq expects no insertions or deletions relative to the reference sequence provided. Indeed, any read with a detected insertion or deletion is automatically discarded during QC. This works well for speeding up analysis of returned reads, but can lead to problems if (1) you expect insertions and deletions or (2) the best-scoring alignment of a read to the reference is one that opens a gap. There are currently no workarounds for problem 1. Problem 2 can be addressed by tuning the alignment parameters.
Alignment parameters are given as optional arguments (see here); the parameter gap_open_penalty can be raised further to decrease the probability of problem 2 (i.e. score alignments such that those with gaps are scored poorly). Note that we have stress-tested the code with default alignment parameters against ~40,000 random DNA sequences with random mutation positions and found <10 instances of problem 2 (<0.025% of instances; most cases occurred when multiple mutations were placed next to (or near) one another at the end of the evSeq reads). The default evSeq alignment parameters are thus highly robust, but there are situations where they might need to be tuned. We strongly recommend that users evaluate the alignments returned by evSeq to make sure there are no unexpected insertions or deletions. Poor alignments will result in false sequencing negatives – this can be particularly problematic if there are multiple variants in a well, and not all of them are recognized by the alignment (i.e., evSeq fails to recognize that there is a mixed well). As noted, such a situation would be exceedingly rare, but is worth being aware of. In many cases, alignment issues can be easily detected by reviewing both the decoupled and coupled files. To tell if the aligner has included insertions or deletions, (1) look for mutations present in the decoupled file that are not found in the coupled file and (2) look for “#DEAD#” wells that have more reads than the variable_count argument.
Using evSeq from the command line or GUI
Command line
Thanks to setuptools entry_points, evSeq can be accessed from the command line after installation as if it were added to PATH by running:
evSeq refseq folder --OPTIONAL_ARGS ARG_VALUE --FLAGS
where refseq is the .csv file containing information about the experiment described above, and folder is the directory that contains the raw .fastq files (.gz or unzipped) for the experiment.
For information on optional arguments and flags, run
evSeq -h
or see below.
Note: You must be in the environment in which evSeq was installed or this will not be accessible. If you installed the evSeq environment, run
conda activate evSeq
to activate it before running.
GUI
Upon installation, evSeq automatically installs a shortcut onto your Desktop that will launch the evSeq GUI with a double-click. If evSeq was installed in the evSeq environment, the GUI will always run from that environment without you needing to activate it.
The GUI is designed for use by non-programming experts. If you are comfortable with a command line interface, that is the recommended way to use evSeq. If using the GUI, make sure you check the log file after each run to check for warnings or errors encountered. See details on the log file here.
Once opened, you should see a window that looks like this:
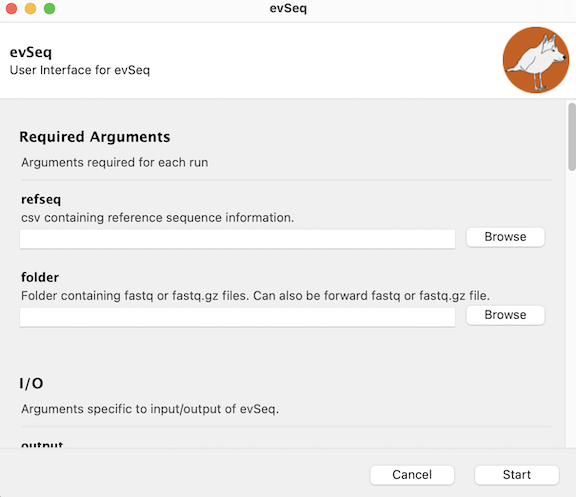
You will see two required arguments — the refseq and folder args — at the top of the GUI. Details on the refseq argument are given below, and the GUI should provide a description of what the folder contains. For more advanced use, other arguments can be accessed by scrolling down. (These additional arguments are detailed in Optional Arguments). You will typically not need these arguments, however, and the standard evSeq run can be started by clicking Start once refseq and folder are populated. Once started, the progress of the program will be printed to the GUI along with any encountered warnings and errors.
Required Arguments
The refseq file
The primary user inputs that are required are contained in the refseq file, which contains information that allows the evSeq software to know how to process each well. From the information contained in this file, evSeq will construct reference sequences for each plate (or well, if using a Detailed refseq file) and analyze the NGS data accordingly.
Default refseq
An example Default refseq format is given in the evSeq GitHub repository here.
This form of the file assumes the same reference sequence in each well of the analyzed plates and requires eight columns: PlateName, IndexPlate, FPrimer, RPrimer, VariableRegion, FrameDistance, BpIndStart, and AaIndStart. These columns are detailed below:
| Column | Type | Description |
|---|---|---|
PlateName |
str |
This is a nickname given to the plate. For instance, if you performed evSeq on a plate that you called “Plate1”, you would put “Plate1” in this column. |
IndexPlate |
DI0X, X=[1,8] |
This is the evSeq index plate used for library preparation corresponding to the plate in PlateName. For instance, if “Plate1” were prepared using index plate 2, IndexPlate would be DI02. Allowed barcode names are DI01 through DI08, as given in the index map file. |
FPrimer |
str, DNA Sequence |
This is the forward inner primer you used to create the amplicon for attaching evSeq barcodes, including the evSeq adapter regions. It should be input exactly as ordered from your oligo supplier, 5’ - 3’. |
RPrimer |
str, DNA Sequence |
This is the reverse inner primer you used to create the amplicon for attaching evSeq barcodes, including the evSeq adapter regions. It should be input exactly as ordered from your oligo supplier, 5’ - 3’. Do not use the reverse complement. |
VariableRegion |
str, DNA Sequence |
This is the entire region between the 3’ ends of each primer used to generate the evSeq amplicon, 5’ - 3’. This is called the “variable region” as it is the sequenced region that can vary meaningfully, since the primers should be invariant. If a specific codon has been specifically mutagenized, this codon may be replaced by “NNN”. See below for more details. |
FrameDistance |
int, [0,1,2] |
Distance (in base pairs) from the 3’ end of FPrimer to the first in-frame codon in your VariableRegion. This is required for accurate translation of sequences. For a given evSeq run, 2 in 3 times the sequence used will be out of reading frame with the full amplicon, so this is important to check. As an example, if the 3’ end of your FPrimer ends on the last base of a codon, your VariableRegion is in-frame and this is argument should be 0. If FPrimer ends on the second base of a codon (e.g., is shifted back 1 bp), then your first in-frame base is 1 base away and this argument should be 1. Note that if “NNN” is used in the VariableRegion, evSeq will double check that you correctly defined this argument — with no “NNN” it will assume you are correct. |
BpIndStart |
int, [0,inf] |
This argument tells the program what index the first base in the variable region belongs to. This is useful for formatting the outputs, as any variation identified in evSeq can be output at the index corresponding to the full gene, rather than the amplicon. |
AaIndStart |
int, [0,inf] |
This argument tells the program what index the first in-frame amino acid in the variable region belongs to. This means that if your FrameDistance argument is not 0, AaIndStart should not be the position of the codon your variable region starts in but rather the next one, since the first codon is not in frame. |
Example Sequence Construction
-------FPrimer------>
5'-AAAAAAAAAAGGGGGGGGGGG-3'
|||||||||||--------VariableRegion------->
5'-CCCCCCCCCCGGGGGGGGGGGTNNNTTTTTTTT...TTTTTTTTTTTTTTTGGGGGGGGGGGGCCCCCCCCCC-3'
FrameDistance = 1 -> x ||||||||||||
3'-CCCCCCCCCCCCAAAAAAAAAA-5'
<-------RPrimer-------
In this simple example, the FPrimer sequence is AAAAAAAAAAGGGGGGGGGGG and the RPrimer sequence is AAAAAAAAAACCCCCCCCCCCC, with the A regions corresponding to the evSeq adapter regions and the G/C regions corresponding to the directional sequence-specific regions. The VariableRegion is the area between them, TNNNTTTTTTTT...TTTTTTTTTTTTTTT. The NNN sequence specifies the correct reading frame, and the 3’ base of FPrimer is 1 base away from this in-frame codon (indicated with the x), therefore FrameDistance is 1. If the VariableRegion started as base 35 in the gene, BpIndStart would be 35 and AaIndStart would correspond to the position of the NNN codon, which would be position 12 (the first base of this codon is base 36, which is the 12th codon overall).
(Note that if any of your sequences look anything like this example, your library preparation will not work. This is to be interpreted as a simple example only.)
VariableRegion containing “NNN”
(OPTIONAL) You may replace the bases at the known mutagenized positions with “NNN” as the codon. Doing so forces evSeq to return the sequence identified at these positions (e.g., from a site-saturation mutagenesis library), whether or not it matches the parent. If you know where your mutations will occur, this is the recommended way to use evSeq; any off-target mutations not given by “NNN” will still be identified and reported.
evSeq Index Plates
As currently deployed, up to 8 plates (DI01–DI08) can be input in a single evSeq run. No more than 8 rows should thus ever be filled in this form of refseq file.
Detailed refseq
An example Detailed refseq format is given in the evSeq GitHub repository here.
This form of the file allows for a different reference sequence in each well of the analyzed plates, rather than the same reference sequence in every well of a given plate. In addition to the column headers given in Default refseq, this form of the file has a required Well column, enabling specification of a different FPrimer, RPrimer, and VariableRegion for each well in the input plates. As currently deployed, up to 8 plates (DI01–DI08) can be input in a single evSeq run, so no more than 768 rows should ever be filled in this form of refseq file.
When using this form of refseq, the detailed_refseq flag must be set (see next sections for details).
folder
This is the folder containing the fastq or fastq.gz files generated during next-gen sequencing. Once activated, evSeq will…
- Look in this folder to find all filenames containing
_R1_or_R2_. - Match forward and reverse files by the name preceding the identified
_R1_or_R2_. For instance, the filesCHL1_S193_L001_R1_001.fastq.gzandCHL1_S193_L001_R2_001.fastq.gzwould be matched because the text preceding the_R1_and_R2_,CHL1_S193_L001, matches for both files. The file with the_R1_is designated the forward read file and the file with the_R2_is designated the reverse read file.
Note that both files without a _R1_ or _R2_ in their name and files for which no matching partner is identified will be ignored; all ignored files are recorded in the log file. If multiple forward-reverse file pairs are identified, evSeq will raise an error.
In special cases the forward read file can be passed in as the folder argument and the reverse read file can be passed in as the optional argument fastq_r. See the entry on fastq_r in the Optional Arguments section for more detail.
Optional Arguments
There are a number of flags and optional arguments that can be passed for evSeq, all detailed in the table below:
| Argument | Type | Description |
|---|---|---|
| Input/Output | ||
fastq_r |
Argument | This argument is only available for command line use. If a case arises where, for whatever reason, evSeq cannot auto-identify the forward and reverse read files, this option acts as a failsafe. Instead of passing the folder containing the forward and reverse files in to the folder required argument, pass in the forward read file as the folder argument and the reverse read file as this optional argument. |
output |
Argument | By default, deSeq will save to the current working directory (command line) or the evSeq Git repository folder (GUI). The default save location can be overwritten with this argument. |
detailed_refseq |
Flag | Set this flag (check the box in the GUI) when passing in a detailed reference sequence file. See Detailed refseq for more information. |
analysis_only |
Flag | Set this flag (check the box in the GUI) to only perform Q-score analysis on the input fastq files. The only output in this case will be the quality score histograms. |
only_parse_fastqs |
Flag | Set this flag to stop evSeq after generation of parsed, well-filtered fastq files. Counts, platemaps, and alignments will not be returned in this case. Used in case the well-specific fastq sequences are desired but not the entire evSeq analysis. |
keep_parsed_fastqs |
Flag | Set this flag to save parsed, well-filtered fastq files as in only_parse_fastqs but to also finish the regular evSeq run. |
return_alignments |
Flag | Set this flag to return alignments along with the evSeq output. Note that this flag is ignored if either analysis_only or stop_after_fastq are used. |
| Read Analysis | ||
average_q_cutoff |
Argument | During initial sequencing QC, evSeq will discard any sequence with an average quality score below this value. The default value is 25. |
bp_q_cutoff |
Argument | Bases with a q-score below this value are ignored when counting the number of sequences aligned at each position. For the coupled outputs (see below), counts are only returned if all bases in the combination pass. The default value is 30. |
length_cutoff |
Argument | During initial sequencing QC, evSeq will discard any sequence with an read with total length below length_cutoff * read_length. The default value is 0.9. |
match_score |
Argument | When making an alignment, matching bases add this value to the score. The default value is 1. |
mismatch_penalty |
Argument | When making an alignment, mis-matching bases subtract this value from the score. The default value is 0. |
gap_open_penalty |
Argument | When making an alignment, opening a gap subtracts this value from the score. The default value is 3. |
gap_extension_penalty |
Argument | When making an alignment, extending a gap subtracts this value from the score. The default value is 1. |
| Position Identification | ||
variable_thresh |
Argument | This argument sets the threshold that determines whether or not a position is variable. In other words, if a position contains a non-reference sequence sequence at a given position at a fraction greater than variable_thresh, then it is a variable position. The default is 0.2. Setting this value lower makes evSeq more sensitive to variation, while setting it higher makes it less sensitive. A value of 1, for instance, would find no variable positions. |
variable_count |
Argument | This sets the count threshold for identifying “dead” wells. If a well has fewer sequences that pass QC than this value, then it is considered “dead”. The default value is 10 (meaning only wells with fewer than 10 sequences are dead). |
| Advanced | ||
jobs |
Argument | This is the number of processors used by deSeq for data processing. By default, evSeq uses 1 less processor than are available on your computer. As with all multiprocessing programs, it is typically not recommended to use all available processors unless you are okay devoting all computer resources to the task (e.g. you don’t want to be concurrently checking email, playing music, running another program, etc.). The number of jobs can be lowered to reduce the memory demands of evSeq. |
read_length |
Argument | By default, evSeq will attempt to determine the read length from the fastq files. If this process is failing (e.g., due to heavy primer-dimer contamination), the read length can be manually set using this argument. |
fancy_progress_bar |
Flag | Launches a tqdm.gui instance for intensive evSeq processes to give you a better estimate for performance/time remaining on your run. While tqdm.gui is still in experimental/alpha stages (which it will warn you about), we have not found any problems with this as of yet. |
Next page: Understanding the outputs.
Back to the main page.