evSeq
No sequence-function pair left behind.
Every Variant Sequencing (evSeq) is a library preparation and analysis protocol that slots neatly into existing workflows to enable extremely low-cost, massively parallel sequencing of protein variants. Designed for heterologously expressed protein variants arrayed in 96-well plates (or similar), this workflow enables sequencing all variants from targetted mutagenesis libraries produced during a protein engineering or biochemical mutagenesis experiment at a cost of cents per variant, even for labs that do not have expertise in or access to next-generation sequencing (NGS) technology.
Read the Paper!
This repository accompanies the work “evSeq: Cost-Effective Amplicon Sequencing of Every Variant in Protein Mutant Libraries”.
Read the Docs!
Find detailed documentation at the individual pages linked below or start at the overview.
Quick links to common resources:
Biology
Computation
Troubleshooting
General Overview
The evSeq workflow
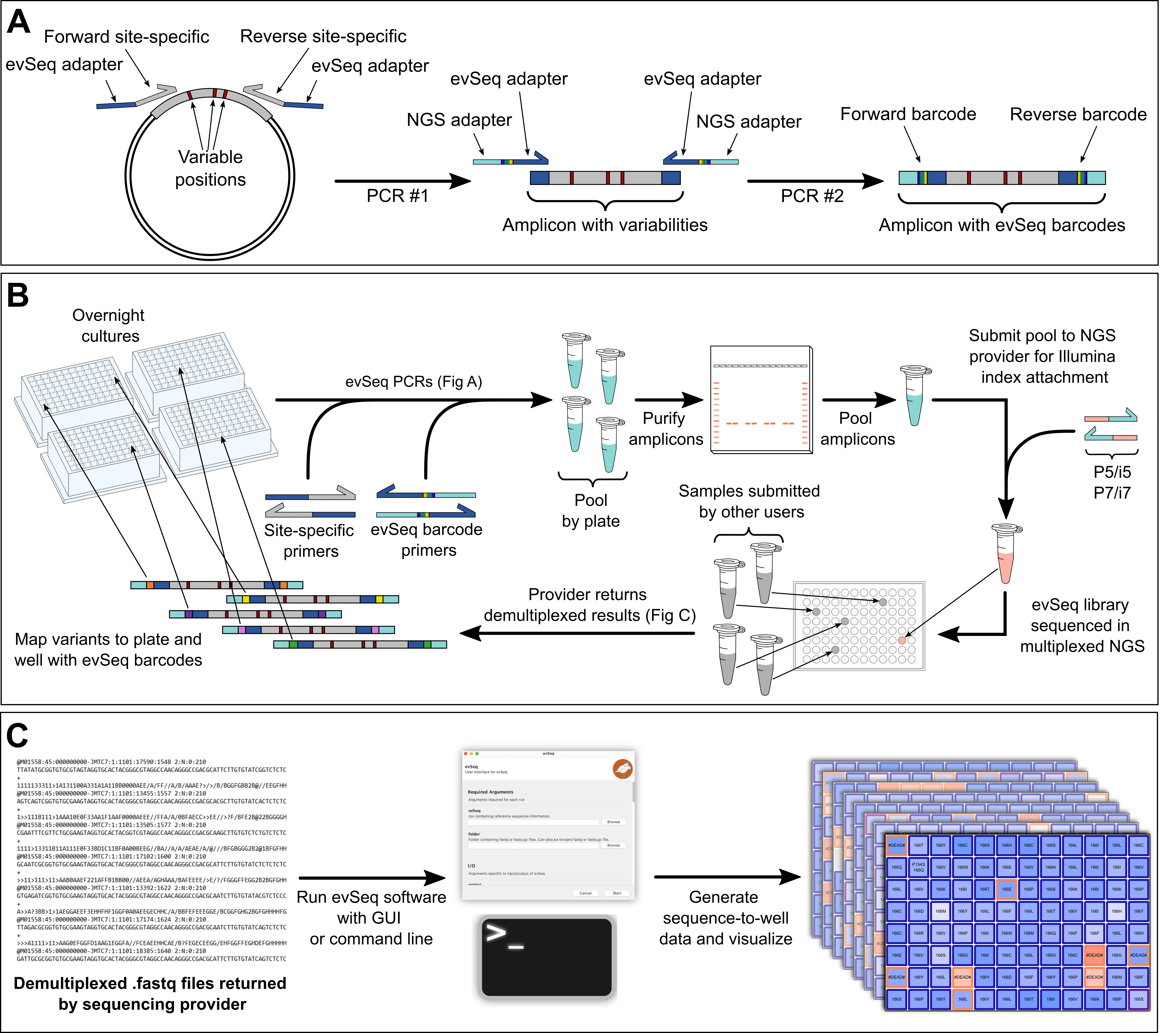 A)
A) evSeq amplifies out a region of interest that contains variability, attaches well-specific barcodes and adapters, and is ready for NGS.
B) All that’s required to perform the evSeq laboratory procedure is:
- a 96-well thermalcycler
- standard PCR reagents and materials
- access to an NGS provider
- two 96-well plates of
evSeqbarcoding (“outer”) primers - a pair of region-specific
evSeq-compatible (“inner”) primers - 96-well plate(s) of cultures containing DNA encoding protein variants
- a 12-channel 10 µL pipette is also helpful
That’s it.
Due to the two-primer, culture-based PCR methodology employed by evSeq, only a new pair of inner primers needs to be ordered when targeting new regions/sequences and no DNA isolation needs to be performed.
C) Once the sequences are returned by the NGS provider, the computational workup can be performed on a standard laptop by users with little-to-no computational experience.
The amplicons prepared with evSeq can yield nearly 1000 high-quality protein variant sequences for just the cost of the multiplexed NGS run (typically ~$100 from commercial sequencing providers, likely lower for in-house providers).
Construct and visualize sequence-function pairs
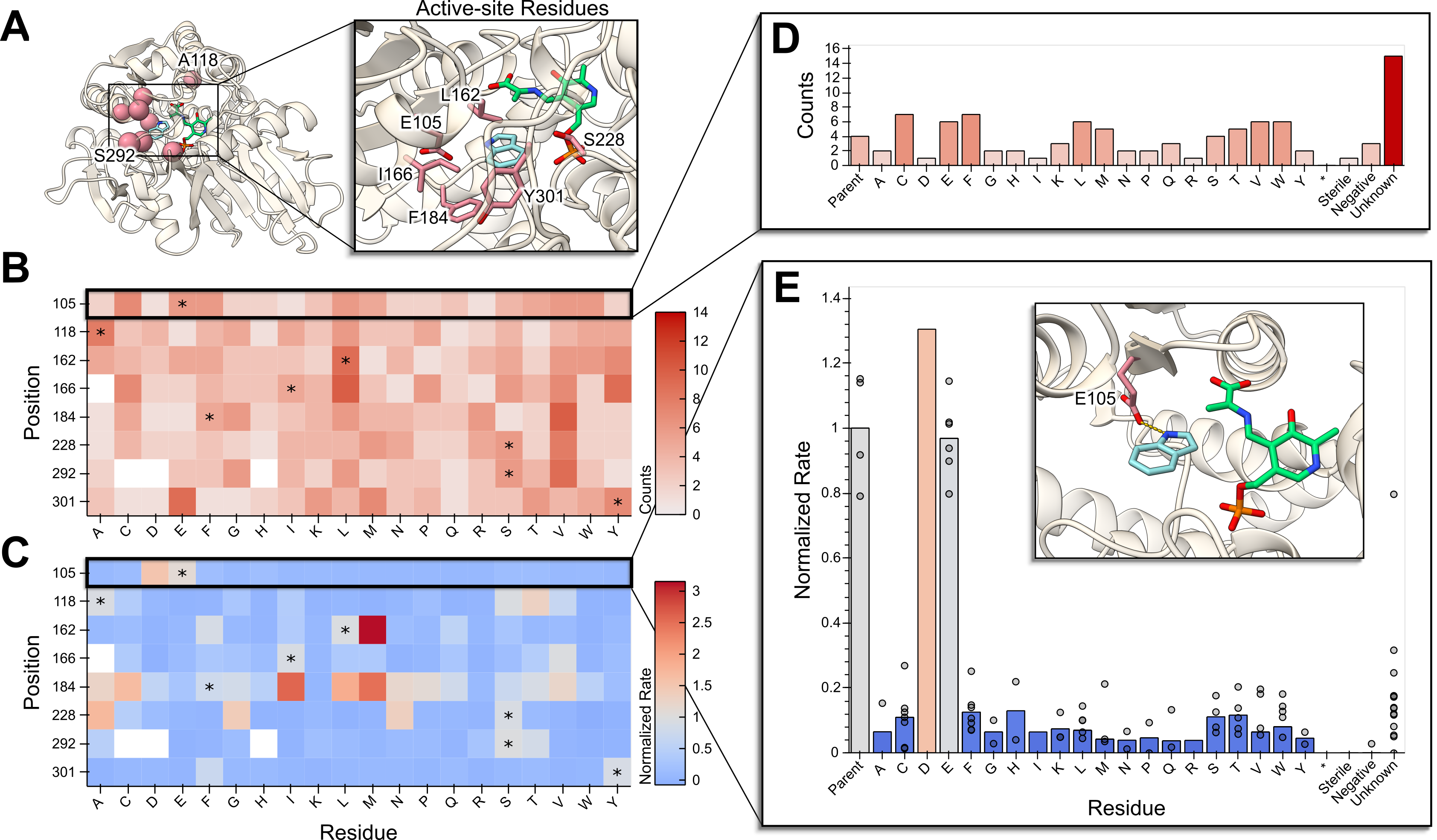 Sequencing eight site-saturation libraries (768 wells) in a single
Sequencing eight site-saturation libraries (768 wells) in a single evSeq run and combining this with activity data to create low-cost sequence-function data. A) Enzyme and active-site structure highlighting mutated residues. B) Heatmap of the number of identified variants/mutations (“counts”) for each position mutated (“library”) from processed evSeq data. C) Heatmap of the average activity (“normalized rate”) for each variant/mutation in each library. D) Counts for a single library, also showing the number of unidentified wells. E) Activity for a single library, showing biological replicates. (Inset displays the mutated residue in this library.)
Documentation
Biology
Theoretical overview
Library preparation
- Dual-Index Barcode Plates
- Inner Primer Design
- Inner Primer Test PCR
- PCR Protocol
- PCR Product Purification
Computation
Computational basics
Installation
Running evSeq
Understanding the Outputs
Additional Examples
Below are a collection of Jupyter Notebooks (rendered as documents) with examples on how to get the most out of evSeq. If you want to run them on your own, they can be found in the examples directory of the evSeq repository.
Using evSeq data
- Importing and viewing
evSeqdata - Pairing sequence to function
- Analyzing single-site-saturation libraries
evSeqfor multisite libraries- Generating our visualizations
- Submitting to Protαβank and other databases
- Miscellaneous visualizations
Creating barcode/index pairs
Running evSeq in a Jupyter Notebook
- To run this notebook on your own, open it in the
evSeqrepository and run it from its current location (found as evSeq/examples/8-full_demo.ipynb).
Troubleshooting
- Poor reverse read quality
- Poor results but good quality sequencing
- Progress bar not showing up in Jupyter
- Windows:
CommandNotFoundError: Your shell has not been properly configured to use 'conda activate' - Windows: The GUI Will Not Open
- macOS:
PermissionError: [Errno 1] Operation not permitted - macOS:
xcrunError - Linux:
CondaEnvException: Pip failed